上月的文獻(xiàn)學(xué)習(xí),我們梳理了一篇影響因子7分左右的關(guān)于乙酰化的SCI,文章涉及乙酰化研究的實(shí)驗(yàn)不多,是不是意猶未盡呢?今天,我們一起來(lái)看看影響因子16左右的文章怎么研究乙酰化的吧~
這次分享的文獻(xiàn)題目是:KAT6A Acetylation of SMAD3 Regulates Myeloid-Derived Suppressor Cell Recruitment, Metastasis, and Immunotherapy in Triple-Negative Breast Cancer,影響因子為16.803分,兩個(gè)月前才熱乎乎的發(fā)表在Adv Sci雜志上。
想了解高分文章是怎么研究乙酰化的伙伴們,現(xiàn)在就跟著我,踏著之前文獻(xiàn)解讀的“標(biāo)準(zhǔn)步驟”往下看吧~
一、KAT6A的過(guò)表達(dá)與三陰性乳腺癌(TNBC)轉(zhuǎn)移相關(guān)
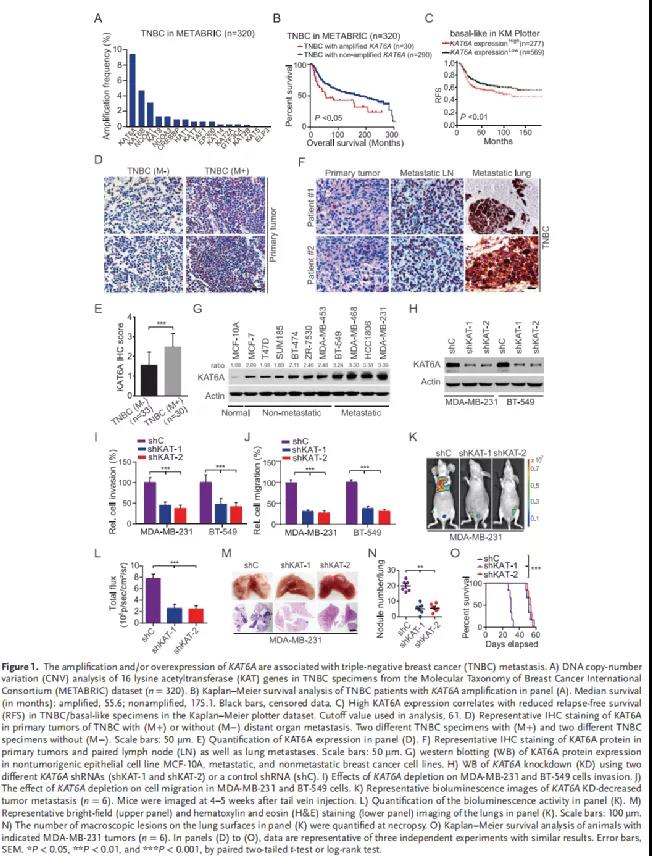
為了確定KAT6A在乳腺癌轉(zhuǎn)移中的作用,作者首先用METABRIC dataset 分析了16個(gè)賴氨酸乙酰轉(zhuǎn)移酶(KAT)基因DNA拷貝數(shù)的改變情況。結(jié)果顯示,KAT基因在TNBC中表現(xiàn)出不同程度的拷貝數(shù)擴(kuò)增,而KAT6A拷貝數(shù)擴(kuò)增最多(Figure 1A)。Kaplan-Meier分析發(fā)現(xiàn)KAT6A擴(kuò)增拷貝數(shù)高的TNBC患者,總生存期更差(Figure 1B)。此外,高表達(dá)的KAT6A亦與無(wú)復(fù)發(fā)生存率的降低顯著相關(guān)(Figure 1C)。接下來(lái)的免疫組化結(jié)果顯示遠(yuǎn)處轉(zhuǎn)移的TNBC腫瘤KAT6A表達(dá)明顯高于無(wú)腫瘤轉(zhuǎn)移的TNBC(Figure 1C-1D)。與原發(fā)性TNBC腫瘤相比,KAT6A在淋巴結(jié)和肺轉(zhuǎn)移中均顯著上調(diào)(Figure 1F)。細(xì)胞水平檢測(cè)發(fā)現(xiàn),與MCF-10A細(xì)胞相比,KAT6A在所有檢測(cè)的乳腺癌細(xì)胞中都有高表達(dá),具有高轉(zhuǎn)移能力的TNBC細(xì)胞中KAT6A蛋白水平高于非轉(zhuǎn)移細(xì)胞(Figure 1G)。在MDA-MB-231和BT-549細(xì)胞中敲低KAT6A(Figure 1H)顯著降低了細(xì)胞的增殖能力(補(bǔ)充材料),侵襲和遷移能力(Figure 1I - 1J)以及原位異種移植瘤的生長(zhǎng)和轉(zhuǎn)移能力(Figure 1K-1L)。同時(shí),KAT6A的敲低亦可顯著抑制轉(zhuǎn)移性肺結(jié)節(jié)的形成(Figure 1M- 1N)、延長(zhǎng)動(dòng)物的生存期(Figure 1O)。這些數(shù)據(jù)表明,KAT6A對(duì)TNBC轉(zhuǎn)移至關(guān)重要,KAT6A高表達(dá)的患者預(yù)后較差。二、KAT6A使SMAD3在Lys20和Lys117發(fā)生乙酰化
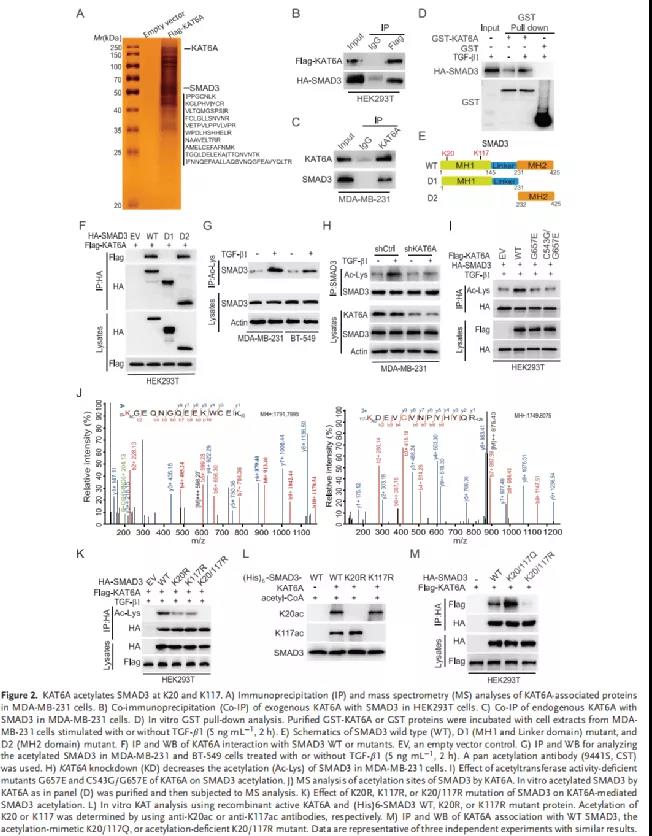
為了探索KAT6A在乳腺癌轉(zhuǎn)移中調(diào)控的下游靶點(diǎn),作者從MDA-MB-231細(xì)胞中純化了flag標(biāo)記的KAT6A復(fù)合物,并進(jìn)行MS分析。在鑒定出KAT6A可結(jié)合蛋白中,SMAD3高度富集(Figure 2A)。外源性以及內(nèi)源性Co-IP分析均證實(shí)KAT6A與SMAD3的存在相互作用(Figure 2B-2C)。GST pulldown實(shí)驗(yàn)發(fā)現(xiàn)純化的重組KAT6A可直接與SMAD3相互作用,TGF-β1的刺激顯著增強(qiáng)KAT6A-SMAD3的關(guān)聯(lián)(Figure 2D)。在HEK293T細(xì)胞中,KAT6A與野生型SMAD3或缺失突變體SMAD3共表達(dá)分析發(fā)現(xiàn):SMAD3的c端MH2結(jié)構(gòu)域(氨基酸232-425)是與KAT6A關(guān)聯(lián)所必需的(Figure 2E-2F)。接下來(lái),作者評(píng)估了KAT6A 敲低對(duì)SMAD3乙酰化的影響。TGF-β1激活SMAD3的乙酰化(Figure 2G),而KAT6A的敲低減弱了TGF-β1誘導(dǎo)的SMAD3乙酰化(Figure 2H)。與野生型 KAT6A相比,KAT6A乙酰轉(zhuǎn)移酶活性缺陷突變體G657E或C543G/G657E顯著減弱了SMAD3的乙酰化(Figure 2I)。
質(zhì)譜分析發(fā)現(xiàn)SMAD3 MH1區(qū)域賴氨酸20(K20)和賴氨酸117(K117)發(fā)生乙酰化,同時(shí),這兩個(gè)殘基在不同物種間高度保守(Figure 2J)。在TGF-β1刺激下,賴氨酸到精氨酸的突變降低KAT6A誘導(dǎo)的SMAD3乙酰化,而賴氨酸和精氨酸的同時(shí)突變消除SMAD3的乙酰化(Figure 2K)。使用純化的重組活性KAT6A和重組WT SMAD3或K20R或K117R突變體進(jìn)行體外KAT實(shí)驗(yàn)證實(shí)KAT6A對(duì)K20和K117的乙酰化(Figure 2L)。此外,與WT SMAD3相比,乙酰化模擬K20/117Q突變體增強(qiáng)了SMAD3與KAT6A的關(guān)聯(lián),而非乙酰化K20/117R突變體抑制了它們的相互作用(Figure 2M)。
因此,這些數(shù)據(jù)表明,KAT6A可以直接乙酰化SMAD3的K20和K117殘基。
三、SMAD3的K20/K117乙酰化上調(diào)免疫反應(yīng)相關(guān)細(xì)胞因子
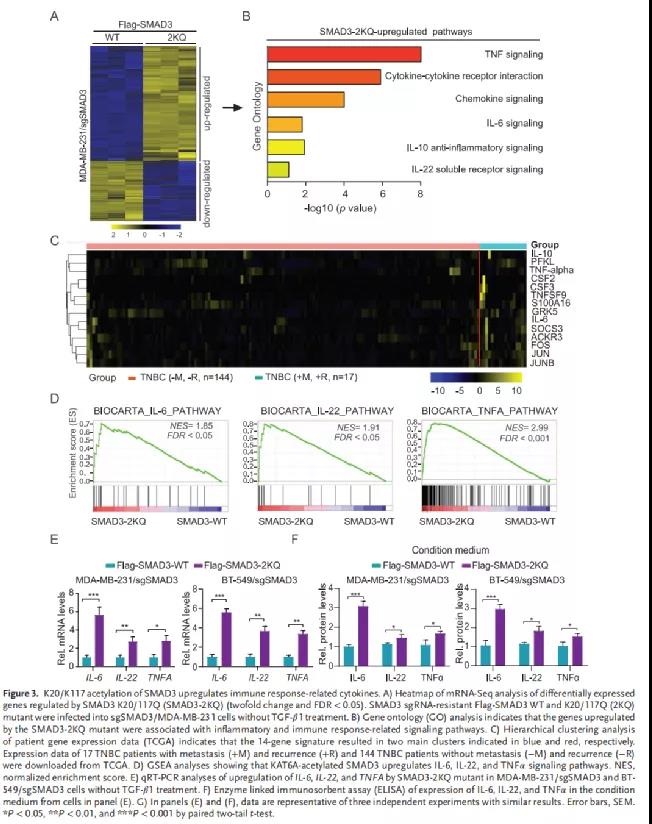
鑒于SMAD3是一個(gè)關(guān)鍵轉(zhuǎn)錄因子,作者假設(shè)SMAD3的K20/K117乙酰化增強(qiáng)了其轉(zhuǎn)錄活性。為驗(yàn)證這一假設(shè),在MDA-MB-231/sgSMAD3細(xì)胞中進(jìn)行了RNA-Seq,這些細(xì)胞過(guò)表達(dá)Flag-SMAD3 WT或精氨酸賴氨酸共同突變體(2KQ突變體)。測(cè)序分析出554個(gè)在2KQ突變體組高表達(dá)的基因(Figure 3A)。這554個(gè)基因在與炎癥和免疫反應(yīng)相關(guān)信號(hào)通路中高度富集(Figure 3B)。接下來(lái),結(jié)合TCGA數(shù)據(jù)庫(kù),作者篩選出無(wú)轉(zhuǎn)移和復(fù)發(fā)的TNBC患者樣本中高表達(dá)的14個(gè)基因(Figure 3C)。基因集富集分析顯示,KAT6A乙酰化的SMAD3(SMAD3-2KQ)上調(diào)炎癥和免疫反應(yīng)相關(guān)的(IL-6、IL-22和TNFα)信號(hào)通路(Figure 3D)。此外,QPCR分析發(fā)現(xiàn),2KQ突變體促進(jìn)了MDA-MB-231/sgSMAD3和BT-549/sgSMAD3細(xì)胞中IL-6、IL-22和TNFα的表達(dá)和分泌(Figure 3E-3F)。補(bǔ)充材料中,與SMAD3-WT相比,SMAD3-2KQ突變體顯著提高了IL-6、IL-22和TNFA啟動(dòng)子的活性。ChIP-qPCR分析發(fā)現(xiàn)MDA-MB和BT-249細(xì)胞中SMAD3可與IL-6、IL-22和TNFA啟動(dòng)子結(jié)合。TGF-β1刺激確實(shí)可以以KAT6A依賴的方式導(dǎo)致這些細(xì)胞因子表達(dá)的增加。綜上,SMAD3的K20/K117乙酰化上調(diào)免疫應(yīng)答相關(guān)細(xì)胞因子的表達(dá)。四、SMAD3K的20/117位點(diǎn)乙酰化后能增加與KAT6A介導(dǎo)的TRIM24閱讀蛋白的H3K23乙酰化,從而增強(qiáng)SMAD3的活性
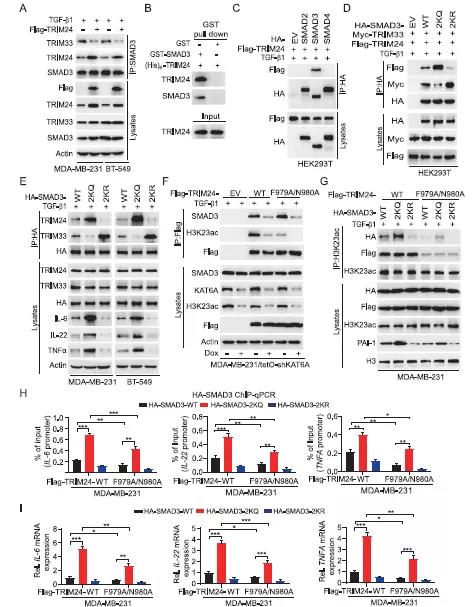
文獻(xiàn)表明,TRIM24是TRIM超家族中TRIM33相關(guān)的輔助因子,也是KAT6A介導(dǎo)的H3K23ac在癌癥中的閱讀蛋白。因此作者研究了K20/117乙酰化是否能影響SMAD3與TRIM33和/或TRIM24的結(jié)合作用。結(jié)果發(fā)現(xiàn),在TGF-β1刺激的MDA-MB-231和BT-549細(xì)胞中,TRIM24和TRIM33均與SMAD3結(jié)合(Figure 4A-B),且過(guò)表達(dá)TRIM24后,其與SMAD3的結(jié)合增強(qiáng)了,而TRIM33與SMAD3的結(jié)合卻遭到了破壞(Figure 4A)。另外發(fā)現(xiàn),除了SMAD3之外,SMAD2和SMAD4與TRIM24沒(méi)有特異性關(guān)聯(lián)(Figure 4C)。此外,通過(guò)補(bǔ)充材料說(shuō)明TRIM24蛋白的中間段的393-823氨基酸殘基介導(dǎo)了與SMAD3的結(jié)合。過(guò)表達(dá)TRIM33后發(fā)現(xiàn)SMAD3-TRIM24的相互結(jié)合作用減少。以上結(jié)果說(shuō)明TRIM24和TRIM33競(jìng)爭(zhēng)性與TGF-β1激活的SMAD3蛋白相結(jié)合。接下來(lái),作者探索K20/117乙酰化對(duì)SMAD3與TRIM24和TRIM33結(jié)合的影響。與野生型SMAD3相比,乙酰化擬合2KQ突變體增強(qiáng)了TRIM24-SMAD3的相互作用,抑制了TRIM33與SMAD3的結(jié)合。而乙酰化缺失的2KQ突變體則得到了相反的結(jié)果(Figure 4D-4E)。2KQ突變體異位表達(dá)增加了IL-6、IL-22和TGF-β1的蛋白表達(dá)(Figure 4E)。以上結(jié)果表明,SMAD3的乙酰化促進(jìn)了其與致癌基因TRIM24的相互作用,并降低了其與TRIM33腫瘤抑制因子的結(jié)合,從而增強(qiáng)了SMAD3的致癌活性。作者通過(guò)文獻(xiàn)得到假設(shè):KAT6A乙酰化H3K23招募TRIM24-SMAD3復(fù)合物,增強(qiáng)SMAD3-染色質(zhì)相互作用,進(jìn)而導(dǎo)致乳腺癌中SMAD3活化增強(qiáng)。首先,作者采用TRIM24 F979A/N980A的突變體破壞了TRIM24-H3K23ac的交互作用,但對(duì)TRIM24與SMAD3的相關(guān)性沒(méi)有影響(Figure 4F)。在MDA-MB-231細(xì)胞中,共表達(dá)HA-tagged的SMAD3 WT、2KQ或2KR突變體與WT TRIM24 F979A/N980A突變體。結(jié)果得到,與WT TRIM24相比,F(xiàn)979A/N980A突變降低H3K23ac與TRIM24以及SMAD3 WT和突變體的關(guān)聯(lián)(Figure 4G),也降低了SMAD3 WT和2KQ突變體與IL-6、IL-22和TNFα啟動(dòng)子的結(jié)合作用, 從而抑制IL-6、IL-22和TNF-β1的表達(dá)(Figure 4H-4I)。與SMAD3 WT與F979A/N980A突變共轉(zhuǎn)染組相比,2KQ突變顯著增加其與IL-6、IL-22和TNFA啟動(dòng)子的結(jié)合以及增強(qiáng)IL-6、IL-22和TNFA的表達(dá)(Figure 4H-I)。這些結(jié)果表明SMAD3 2KQ突變?cè)黾覫L-6、IL-22和TNFA的表達(dá)與H3K23ac沒(méi)有相關(guān)性,但H3K23ac結(jié)合能進(jìn)一步增加SMAD3 2KQ突變體的活性。綜上,KAT6A乙酰化SMAD3促進(jìn)了SMAD3與H3K23ac閱讀器TRIM24的結(jié)合,KAT6A介導(dǎo)的H3K23ac進(jìn)一步增加了TRIM24/SMAD3-染色質(zhì)關(guān)聯(lián),從而導(dǎo)致SMAD3信號(hào)激活增強(qiáng)。五、SMAD3的K20/K117乙酰化通過(guò)增強(qiáng)CSC和MDSCs招募促進(jìn)TNBC的轉(zhuǎn)移
以上結(jié)果提示,KAT6A/SMAD3軸可能影響TNBC中的乳腺癌干細(xì)胞樣細(xì)胞(BCSC)特性。dox誘導(dǎo)KAT6A的敲降降低了IL-6、IL-22和TNFα 的蛋白表達(dá)、p-STAT3、CSC相關(guān)蛋白SOX2和CD44的表達(dá)、ALDH +亞種群、細(xì)胞成球能力(Figure 5A-5D)。以上實(shí)驗(yàn)結(jié)果能被擬乙酰化SMAD3的異位表達(dá)的2KQ突變體所逆轉(zhuǎn)(Figure 5A-5D)。此外,與對(duì)照組相比,KAT6A KD增加了E-cadherin的表達(dá),降低了N-cadherin、ZEB1、Slug和Snail的表達(dá),但在擬乙酰化SMAD3的異位表達(dá)的2KQ突變體組中未觀察到此變化趨勢(shì)(補(bǔ)充材料)。綜上,KAT6A-SMAD3軸能促進(jìn)TNBC中的BCSC特性。為了研究KAT6A依賴SMAD3乙酰化對(duì)TNBC轉(zhuǎn)移的調(diào)控作用,作者在有或沒(méi)有KAT6A KD的4T1/sgSmad3細(xì)胞中表達(dá)Flag-SMAD3 WT和2KR突變體,從IL-6,IL-22,TNFα的表達(dá)、細(xì)胞增殖、遷移、侵襲、MDSCs招募、肺的微小轉(zhuǎn)移等方面說(shuō)明(補(bǔ)充材料)KAT6A誘導(dǎo)的SMAD3乙酰化能促進(jìn)TNBC的轉(zhuǎn)移。接下來(lái),作者利用4T1小鼠乳腺癌轉(zhuǎn)移模型,研究SMAD3乙酰化對(duì)體內(nèi)MDSCs招募的影響。與未誘導(dǎo)細(xì)胞相比,dox誘導(dǎo)的KAT6A KD降低了IL-6、IL-22和TNFα、p-STAT3的表達(dá)(Figure 5E),KAT6A KD抑制腫瘤生長(zhǎng)、肺轉(zhuǎn)移,延長(zhǎng)小鼠存活期(Figure 5F-5J),抑制MDSCs向轉(zhuǎn)移性肺組織和原位瘤的募集以及轉(zhuǎn)移性肺組織CD4+/CD8+ T細(xì)胞耗竭(Figure 5F-5M+補(bǔ)充材料)。以上結(jié)果均能被乙酰化模擬SMAD3 2KQ突變體所逆轉(zhuǎn)(Figure 5E-5M)。故,KAT6A依賴SMAD3乙酰化通過(guò)MDSCs招募促進(jìn)TNBC轉(zhuǎn)移。為了進(jìn)一步證明乳腺癌細(xì)胞分泌的促炎因子在SMAD3介導(dǎo)的腫瘤轉(zhuǎn)移中的關(guān)鍵作用,作者構(gòu)建了IL-6 KO 4T1細(xì)胞并發(fā)現(xiàn)IL-6 KO抑制了SMAD3-2KQ誘導(dǎo)的MDSCs募集(補(bǔ)充材料)。并在體內(nèi)SMAD3-2KQ-誘導(dǎo)的MDSCs招募實(shí)驗(yàn)中也得到了證實(shí)(補(bǔ)充實(shí)驗(yàn))。以上結(jié)果說(shuō)明促炎因子IL-6亦參與SMAD3介導(dǎo)的腫瘤轉(zhuǎn)移中過(guò)程。綜上,KAT6A誘導(dǎo)的SMAD3乙酰化不僅增強(qiáng)乳腺癌干細(xì)胞樣細(xì)胞特性,同時(shí)通過(guò)持續(xù)產(chǎn)生免疫應(yīng)答相關(guān)細(xì)胞因子誘導(dǎo)MDSCs招募,促進(jìn)乳腺癌的進(jìn)展。<section sty




 2022-02-28 15:34:03
2022-02-28 15:34:03




")

 湘公網(wǎng)安備
湘公網(wǎng)安備